
On December 4-6, 2023, we presented our joint research findings with ENEOS Corporation at IBM Quantum Summit 2023 held in New York. This article will introduce the content of our joint research.
Quantum chemistry computation is considered a promising application area for quantum computers due to the intrinsic quantum nature of the systems being modeled[1]. In the realm of quantum computing, variational quantum algorithms using NISQ (Noisy Intermediate-Scale Quantum) devices are commonly pursued. However, these approaches face several challenges: a substantial number of shots and computational steps are required to maintain results within acceptable statical error margins; the statistical errors mean that the outcomes from actual quantum devices are not variational; and the significant impact of noise on the performance of current quantum devices. These issues are well-known in the field of quantum computing with NISQ devices.
Our proposed QSCI (Quantum Selected Configuration Interaction) method restricts the use of quantum computers to quantum sampling, enabling more accurate energy computations[2].
The QSCI algorithm is as follows:
- Prepare an input state on a quantum computer, while the input state is reasonably optimized by methods such as the Variational Quantum Eigensolver (VQE).
- Measure the input state in the computational basis.
- Select important electronic states. Choose an arbitrary number of frequently measured bit strings from the measured results.
- Use the selected bit strings to construct the subspace Hamiltonian, and then perform exact diagonalization. The obtained energy is guaranteed to be equal to or greater than the exact solution , even with noisy real devices.
We applied the QSCI method to two molecular Hamiltonians (diazene and methane ) and reported our results at the IBM Quantum Summit 2023. It's important to highlight that for the quantum sampling in our calculations, we utilized an actual quantum computer (ibm_algiers).
Diazene ()
We investigated the energy accuracy of VQE calculations for the diazene molecule on a simulator using various ansatzes, in order to determine which type of input state is suitable for QSCI. While the standard Ry ansatz converged to an energy accuracy comparable to Hartree-Fock (HF), the Ry Custom 1 ansatz (hereafter Ry Custom ansatz) achieved an energy accuracy nearly equivalent to that obtained with CASCI(4e,4o).
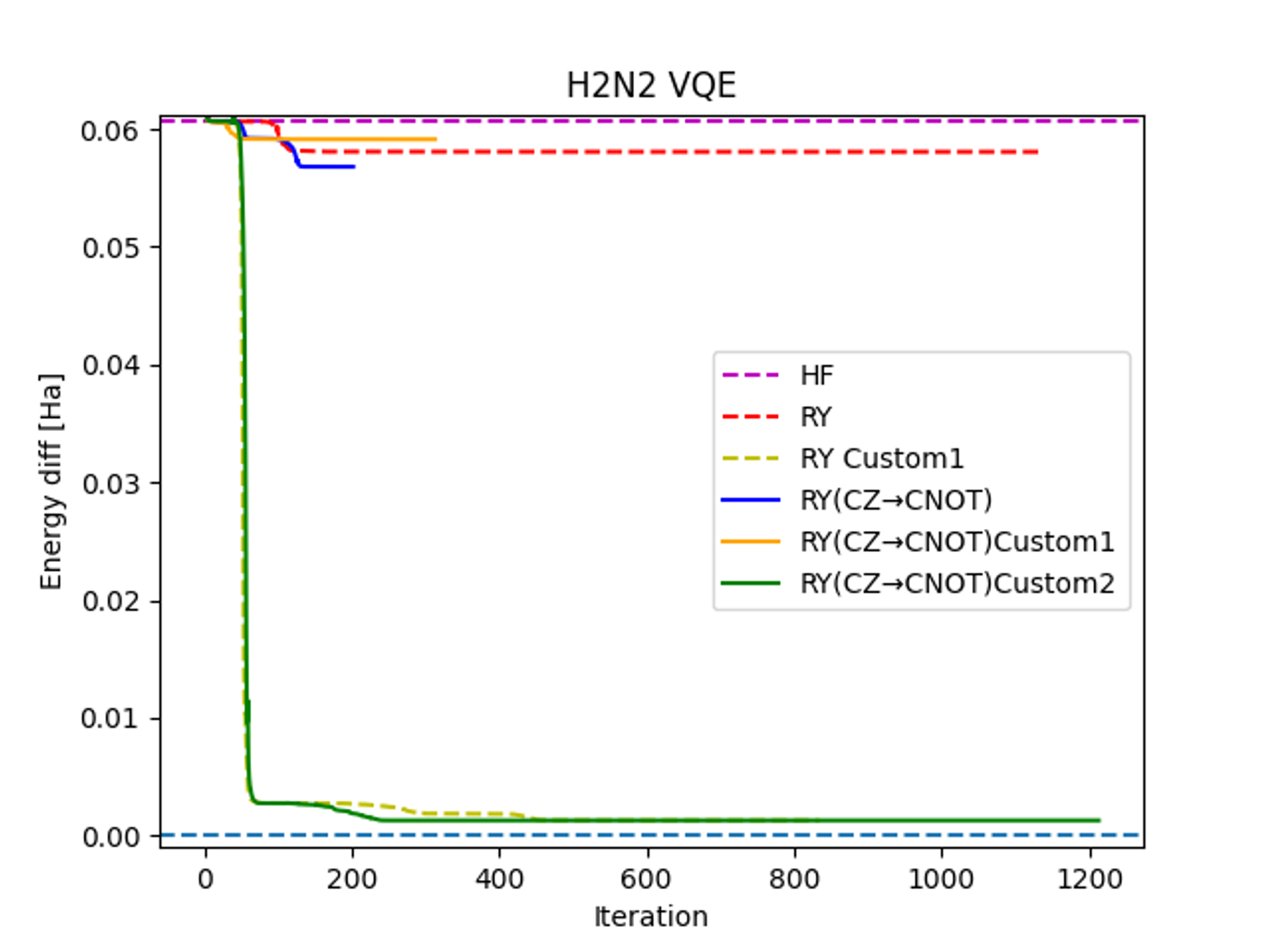
The Ry Custom ansatz involves a modification in the application of CZ gates, as shown in Figure 2. This configuration is achieved by reordering the qubits from the standard Ry ansatz. From a physical perspective, this is equivalent to entangling occupied and virtual orbitals. These findings indicate that the outcomes are significantly influenced not only by the circuit's topology but also by the choice of entanglement pairs.
By inputting the VQE optimized state obtained from the simulator, we conducted real-device QSCI. This is an 8-qubit system, and we utilized ibm_algiers as the actual device. The computational results are presented in Table 1.
Table 1. Ground state energy of the diazene molecule Hamiltonian for various computational methods (in Hartree units). HF: Energy obtained through Hartree-Fock calculation. Input VQE: Optimized energy by using the VQE with Ry Custom ansatz with a depth of 8. Demonstrated QSCI: Energy obtained by executing QSCI using the above state as input. The number of shots was set to 10,000, and an effective Hamiltonian was constructed using all quantum measured states. CASCI(4e,4o): Energy obtained by exact diagonalization within the active space.
| Method | Energy [Ha] |
|---|---|
| HF | -108.5570 |
| Input VQE | -108.6163 |
| Demonstrated QSCI | -108.6176 |
| CASCI(4e,4o) | -108.6176 |
The computational accuracy of QSCI using actual devices was found to surpass that of simulator VQE, and it aligned with more exact calculations (CASCI) with high precision.
Furthermore, we validated the computational performance of real-device QSCI by using simulator VQE results with varying levels of optimization as input states for QSCI (Figure 3).
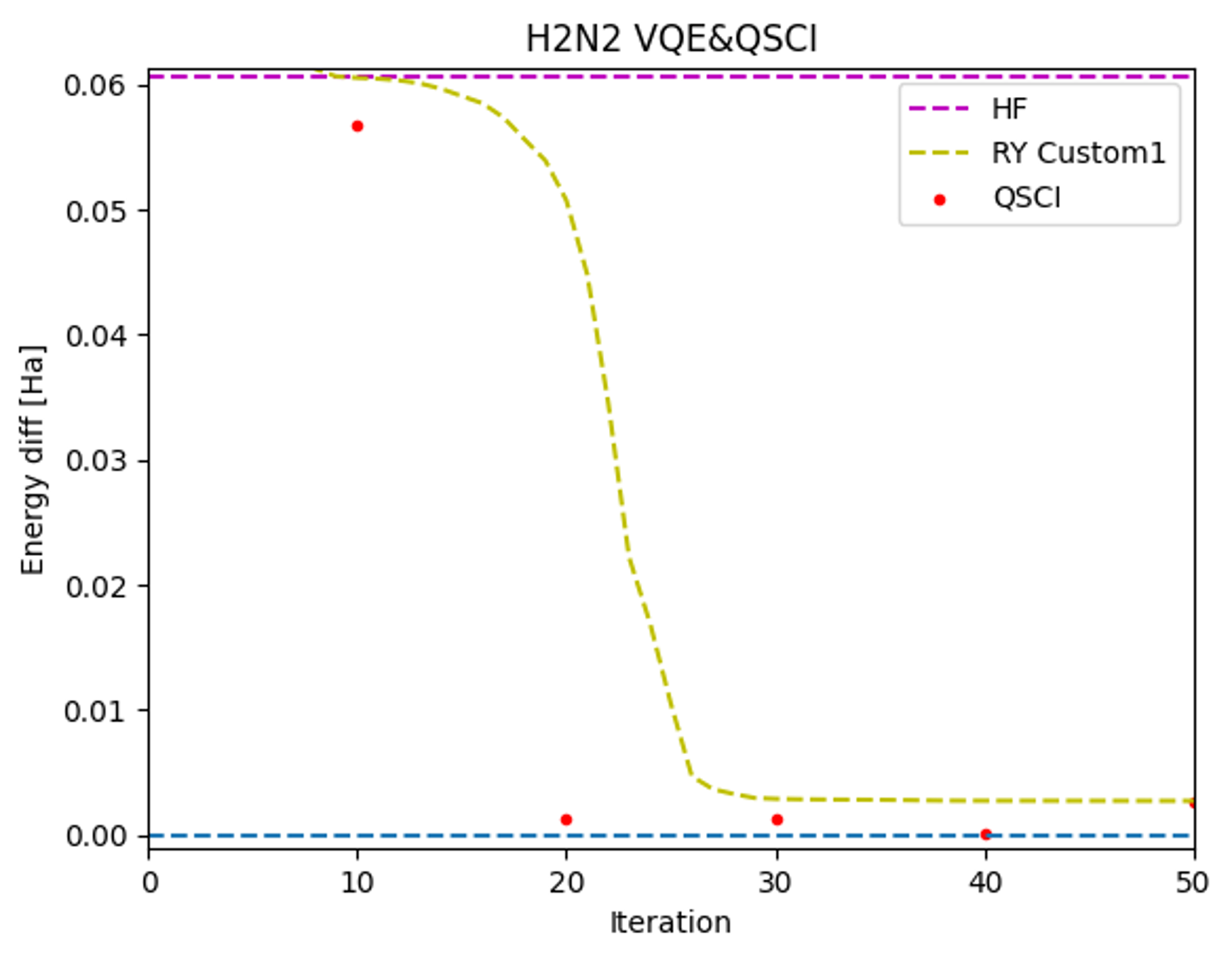
The real-device QSCI consistently demonstrated more accurate computational results than those obtained from the simulator VQE. Notably, high-accuracy ground state energy calculations were possible with real-device QSCI even with VQE input states resulting from around 20 iterations. This ability to accurately calculate energy with QSCI, even from states that are not fully optimized, suggests the potential to overcome one of the challenges of VQE, namely the extensive time required for optimization.
Methane ()
Next, we examined a larger molecular system, methane (, 16-qubit system), specifically focusing on the dissociation reaction of a hydrogen atom:
In our calculations, we modeled this phenomenon by varying the bond length between the hydrogen and carbon atoms, with the most stable state occurring at a bond length of 1.083 Å.
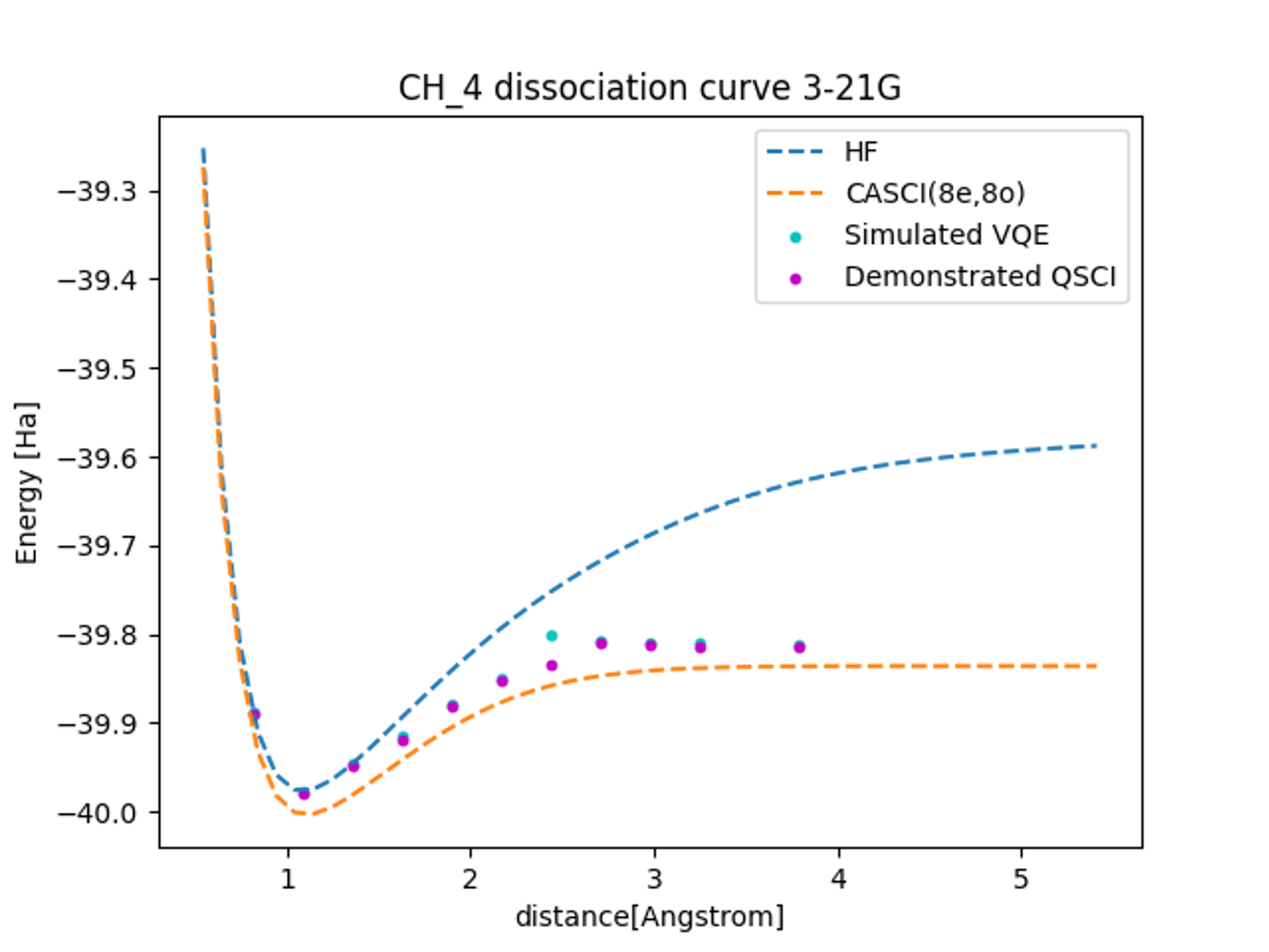
The results of the computations show that the dissociation curves of the ground-state energy for each computational method are depicted in Figure 4. As can be seen from the increasing discrepancy between the Hartree-Fock calculations and the exact solutions with increasing bond length, the contribution of electron correlation becomes crucial in dissociation reactions.
The results of real-device QSCI calculations in a 16-qubit system demonstrate good accuracy compared to noiseless simulator VQE calculations, particularly in the intermediate region. In this study, we only conducted simulations and did not perform VQE calculations on an actual device. However, if we were to carry out real-device calculations with the same 10,000 shots as in the QSCI calculations presented here, we would expect a statistical error of approximately 0.1 Hartree. These results indicate that, even for larger systems compared to other quantum chemistry calculations on quantum hardware, more accurate results can be obtained on actual devices using the QSCI method than VQE.
Conclusion
In this study, we conducted actual quantum computations on a quantum computer (ibm_algiers) for the ground-state energy of two chemical molecules (diazene , methane ). The QSCI method developed by us was applied in the actual computations.
For the diazene molecule (8-qubit):
- The results of the calculations using real-device QSCI were consistent with the exact solutions provided by CASCI(4e,4o).
- The robustness of the QSCI method against the degree of optimization of the input state was demonstrated in real-device computations.
For the dissociation reaction of the methane molecule (16-qubit):
- Real-device quantum computations using the QSCI method achieved results of higher accuracy compared to VQE calculations performed with simulators.
References
[1] S. McArdle, S. Endo, A. Aspuru-Guzik, S. C. Benjamin, and X. Yuan, Reviews of Modern Physics 92, 015003 (2020).
[2] K. Kanno, M. Kohda, R. Imai, S. Koh, K. Mitarai, W. Mizukami, and Y. O. Nakagawa, arXiv: 2302.11320 (2023).
Authors: Yuhei Ikeda, Shoichiro Tsutsui, Keita Kanno